Research Projects
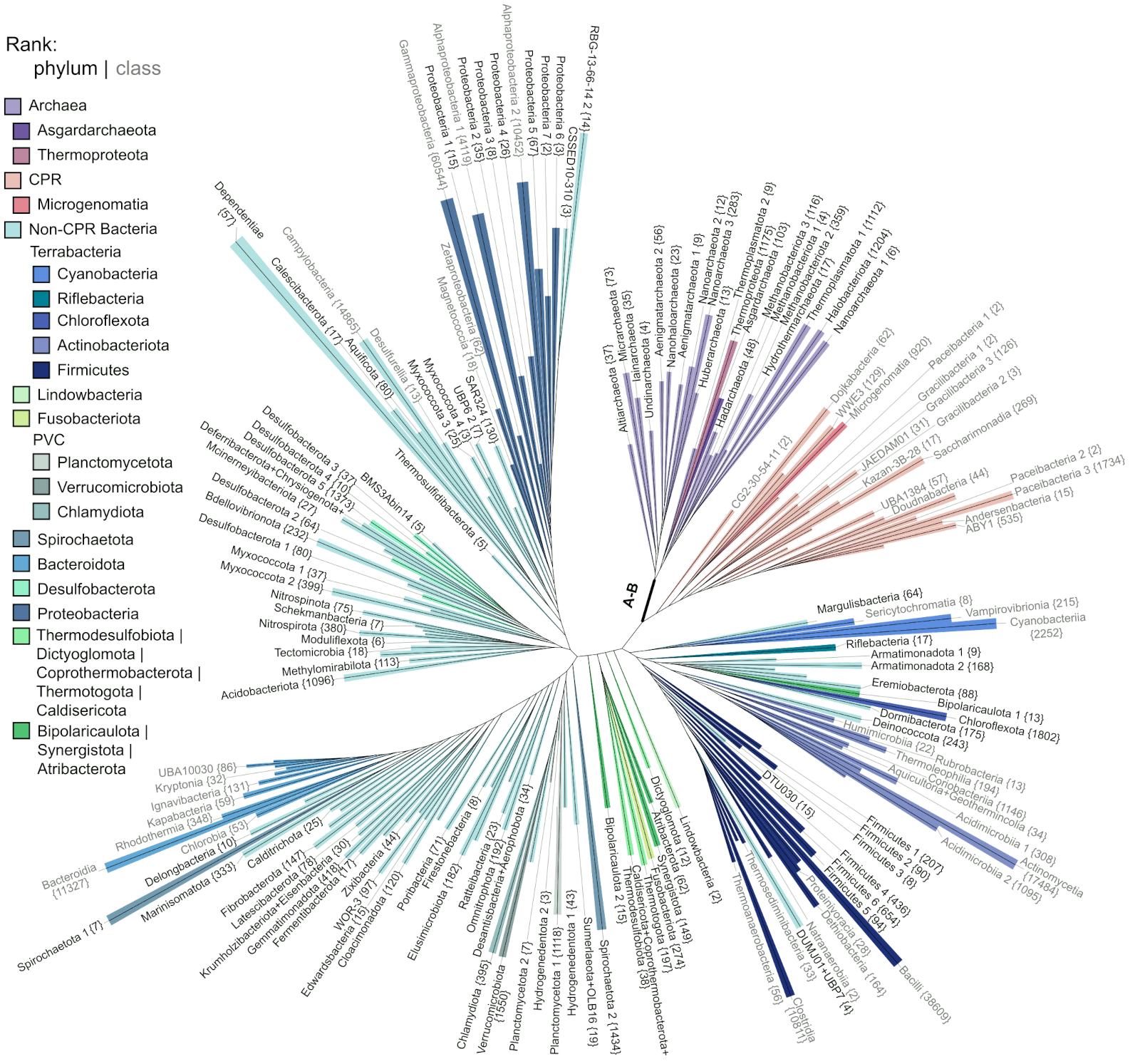
Generation of accurate, expandable phylogenomic trees with uDance (Nature Biotechnology)
Phylogenetic trees provide a framework for organizing evolutionary histories across the tree of life and aid downstream comparative analyses such as metagenomic identification. Methods that rely on single-marker genes such as 16S rRNA have produced trees of limited accuracy with hundreds of thousands of organisms, whereas methods that use genome-wide data are not scalable to large numbers of genomes. We introduce updating trees using divide-and-conquer (uDance), a method that enables updatable genome-wide inference using a divide-and-conquer strategy that refines different parts of the tree independently and can build off of existing trees, with high accuracy and scalability. With uDance, we infer a species tree of roughly 200,000 genomes using 387 marker genes, totaling 42.5 billion amino acid residues. uDance is available publicly at GitHub.
Related work
- Metin Balaban, Yueyu Jiang, Qiyun Zhu et al. Generation of accurate, expandable phylogenomic trees with uDance. Nature Biotechnology, July 2023. https://doi.org/10.1038/s41587-023-01868-8.
- Daniel McDonald, Yueyu Jiang, Metin Balaban et al. Greengenes2 unifies microbial data in a single reference tree. Nature Biotechnology, July 2023. https://doi.org/10.1038/s41587-023-01845-1.
Press coverage
- Efficiently evolving phylogenetic trees: introducing a scalable approach to constructing and updating phylogenies. Link to article.
- New resource harmonizes 16s and shotgun sequencing data for microbiome research. Link to article.
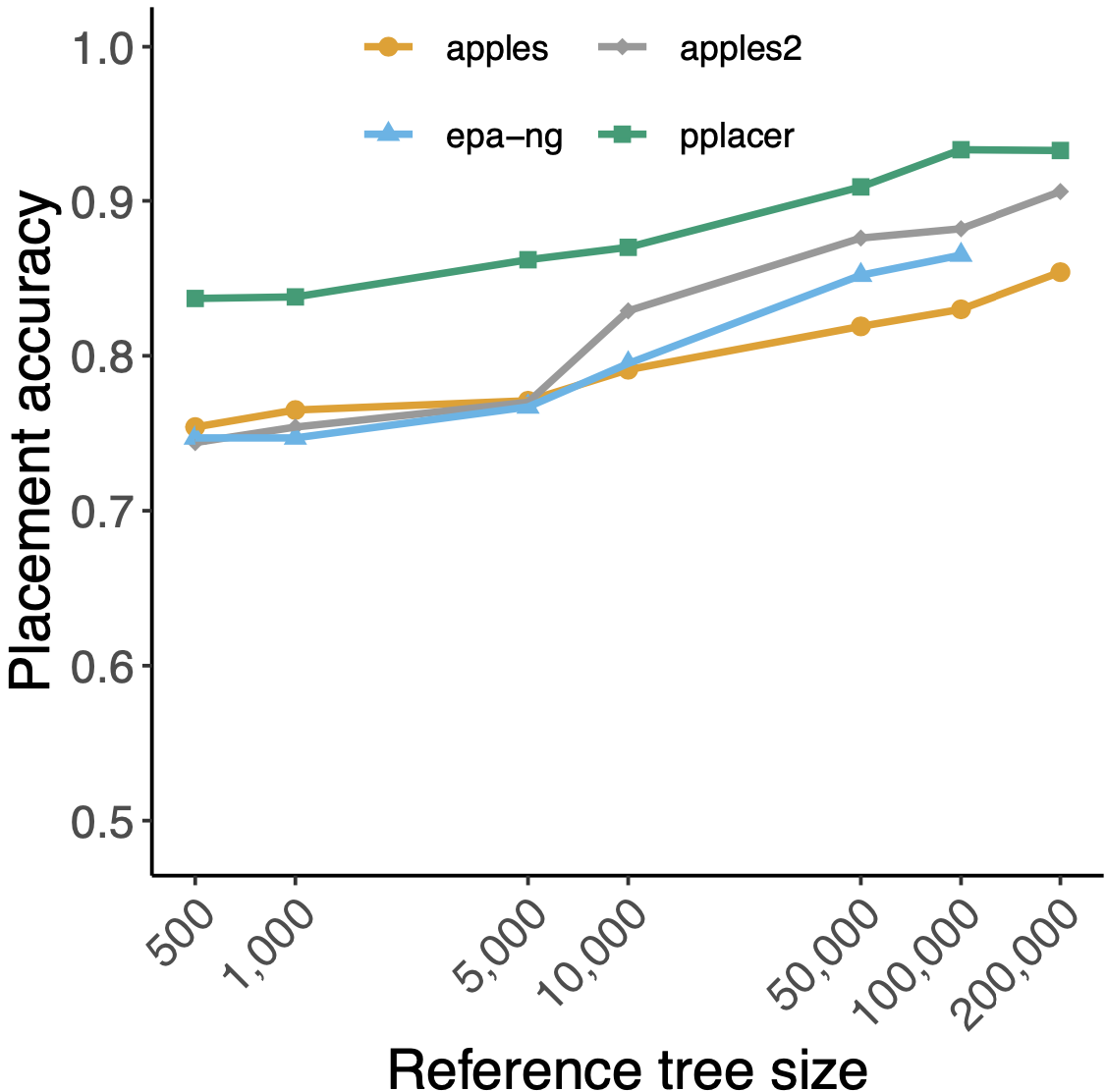
Fast and scalable Distance-Based Phylogenetic Placement
Phylogenetic placement of query samples on an existing phylogeny is increasingly used in microbiome analyses and other biological studies. As the size of available reference trees used in microbiome analyses continues to grow, there is a growing need for methods that place sequences on ultra-large trees (e.g. 200,000 leaves) with high accuracy. In this project, I developed an algorithm called APPLES (and its successor APPLES-2), a distance-based method for phylogenetic placement. In extensive studies, we showed that APPLES-2 is more accurate and scalable, even more so than some of the leading maximum likelihood methods. This scalability is owed to a divide-and-conquer technique that limits distance calculation and phylogenetic placement to parts of the tree most relevant to each query. APPLES-2 is available publicly at GitHub. [Distance-based methods; genome skimming;phylogenetic placement.]
Related work
- Metin Balaban, Shahab Sarmashghi, Siavash Mirarab. APPLES: Scalable Distance-Based Phylogenetic Placement with or without Alignments. Systematic Biology, Volume 69, Issue 3, May 2020, Pages 566–578, https://doi.org/10.1093/sysbio/syz063.
- Metin Balaban, Yueyu Jiang, Daniel Roush, Qiyun Zhu, Siavash Mirarab. Fast and Accurate Distance-based Phylogenetic Placement using Divide and Conquer. Molecular Ecology Resources, 00, 1–15 (2021). https://doi.org/10.1111/1755-0998.13527.
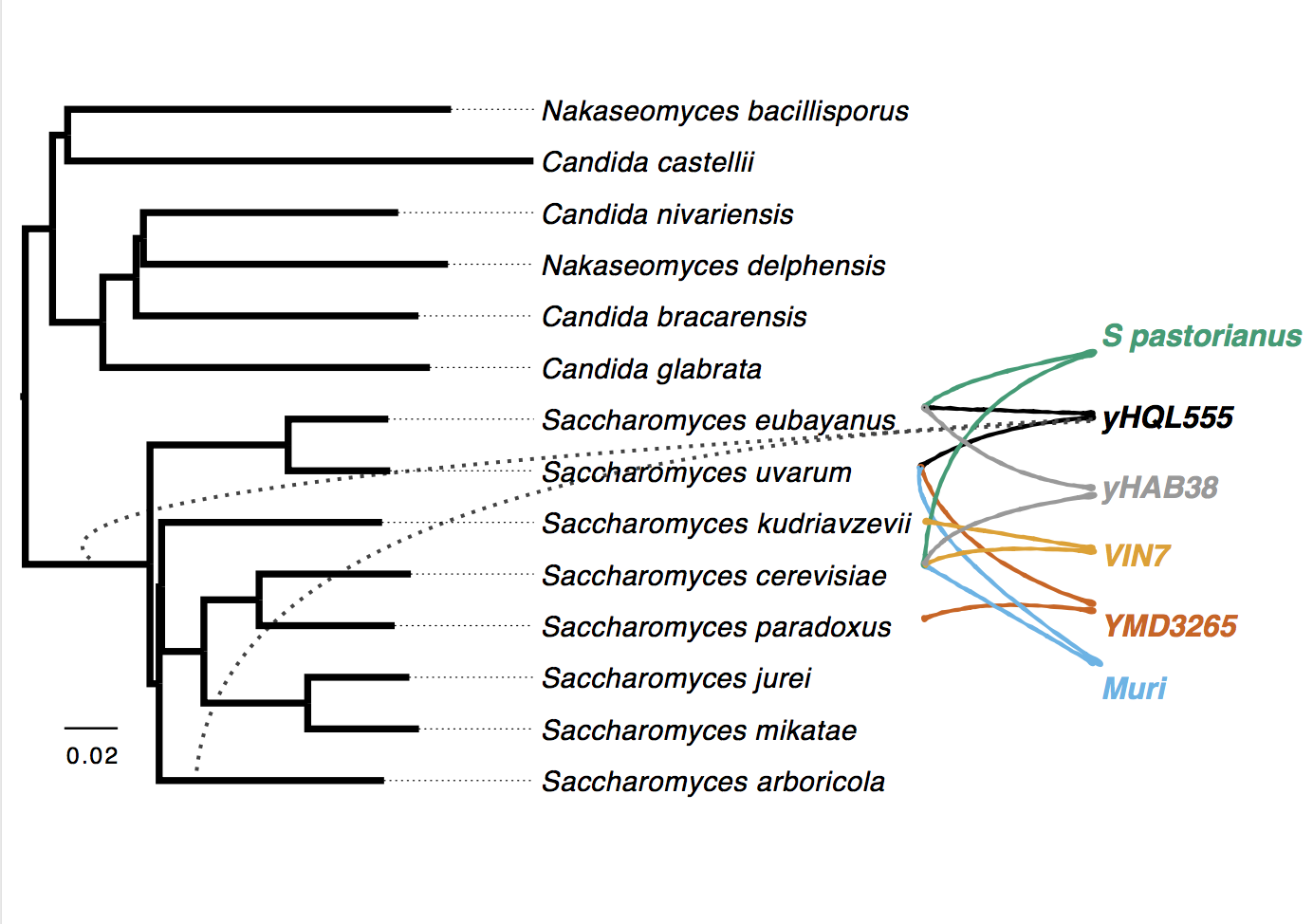
Phylogenetic double placement of mixed samples
Consider a simple computational problem. The inputs are (i) the set of mixed reads generated from a sample that combines two organisms and (ii) separate sets of reads for several reference genomes of known origins. The goal is to find the two organisms that constitute the mixed sample. When constituents are absent from the reference set, we seek to phylogenetically position them with respect to the underlying tree of the reference species. In this project, we introduce a model based on Jaccard indices computed between each sample represented as k-mer sets. The model, built on several assumptions and approximations, allows us to formalize the phylogenetic double-placement problem as a non-convex optimization problem that decomposes mixture distances and performs phylogenetic placement simultaneously. Using a variety of techniques, we are able to solve this optimization problem numerically. Resulting method called MISA and data are available at Github.
Related work
- Metin Balaban, Siavash Mirarab. Phylogenetic double placement of mixed samples. Bioinformatics 36, no. Supplement 1 (2020):i335–43. doi:10.1093/bioinformatics/btaa489.
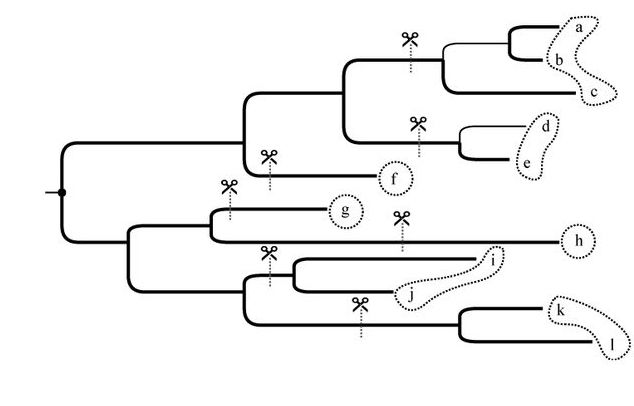
Clustering biological sequences using phylogenetic trees
Clustering homologous sequences based on their similarity is a problem that appears in many bioinformatics applications. The fact that sequences cluster is ultimately the result of their phylogenetic relationships. We defined a family of optimization problems that, given an arbitrary tree, return the minimum number of clusters such that all clusters adhere to constraints on their heterogeneity. These three problems can be solved in time that increases linearly with the size of the tree. We implement these algorithms in a tool called TreeCluster, which we test on three applications: OTU clustering for microbiome data, HIV transmission clustering, and divide-and-conquer multiple sequence alignment. We show that, by using tree-based distances, TreeCluster generates more internally consistent clusters than alternatives and improves the effectiveness of downstream applications. TreeCluster is available at Github.
Related work
- Metin Balaban, Niema Moshiri, Uyen Mai, Xingfan Jia, Siavash Mirarab. TreeCluster: Clustering biological sequences using phylogenetic trees. PLOS ONE 14, no. 8 (2019): e0221068. doi:10.1371/journal.pone.0221068.
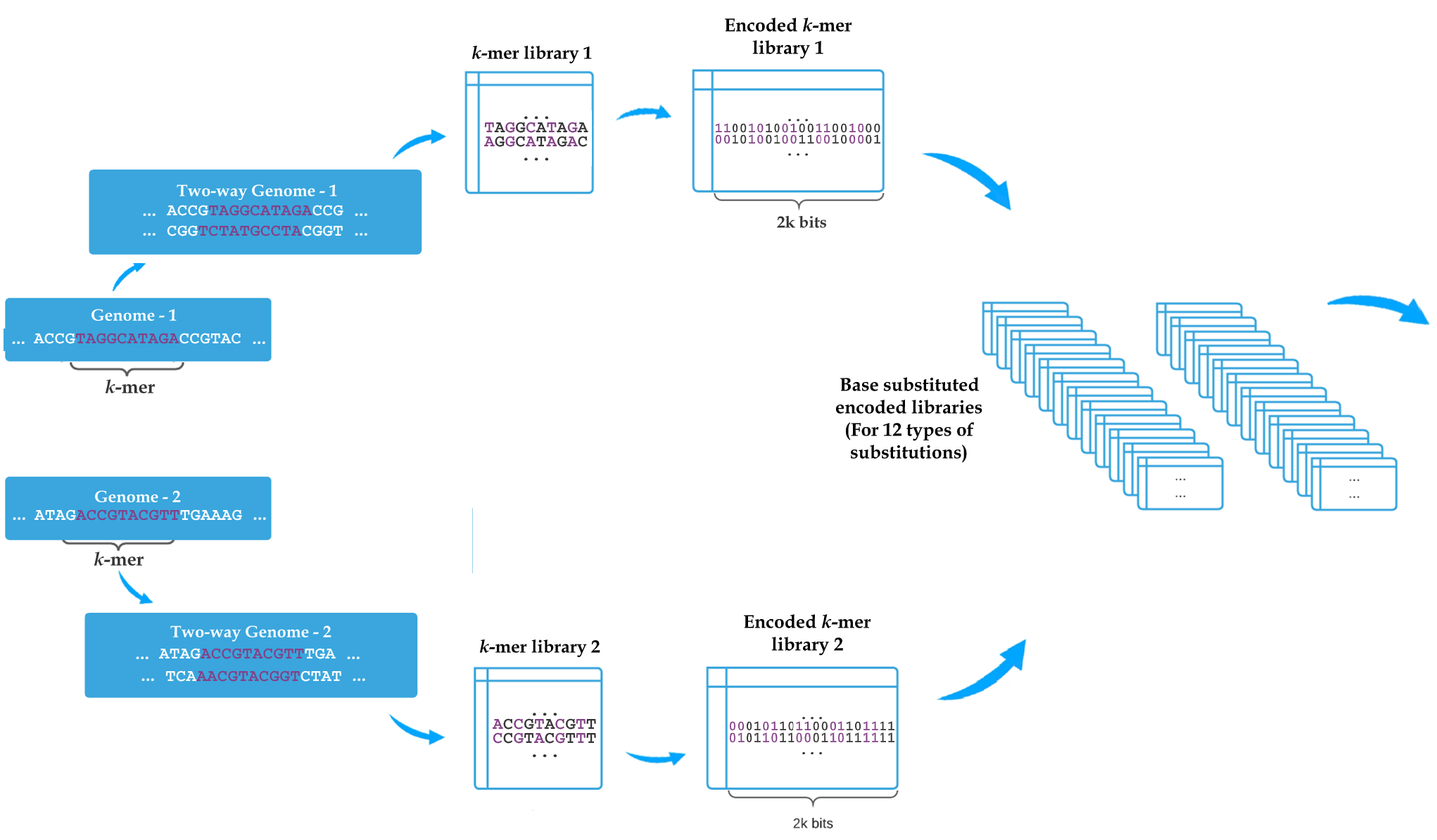
Genome-wide alignment-free distance estimation under a no strand-bias model
The alignment-free methods have much appeal in terms of simplifying the process of inference, especially when analyzing genome-wide data. Despite the appeal, one limitation of alignment-free methods is that they typically rely on simplified models of sequence evolution such as Jukes-Cantor. It is possible to compute pairwise distances under more complex models by computing frequencies of base substitutions provided that these quantities can be estimated in the alignment-free setting. An underappreciated limitation is that for many forms of genome-wide data, which arguably present the best use case for alignment-free methods, the strand of DNA sequences is unknown. Under such conditions, the no-strand bias models are the best that we can do. Here, we show how to calculate distances under a no-strain bias restriction of the General Time Reversible (GTR) model called TK4 without relying on alignments or even assemblies using k-mers.
Related work
- Metin Balaban, Nishat Anjum Bristy, Ahnaf Faisal, Md Shamsuzzoha Bayzid, Siavash Mirarab. Genome-wide alignment-free phylogenetic distance estimation under a no strand-bias model. Bioinformatics Advances, Volume 2, Issue 1, 2022, vbac055. https://doi.org/10.1093/bioadv/vbac055.